Acta. Cryst. (2016) B72(4), 439-459 [ doi:10.1107/S2052520616007447 ] A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom
 The sixth blind test of organic crystal structure prediction (CSP) methods has been held, with five target systems: a small nearly rigid molecule, a polymorphic former drug candidate, a chloride salt hydrate, a co-crystal and a bulky flexible molecule. This blind test has seen substantial growth in the number of participants, with the broad range of prediction methods giving a unique insight into the state of the art in the field.
The sixth blind test of organic crystal structure prediction (CSP) methods has been held, with five target systems: a small nearly rigid molecule, a polymorphic former drug candidate, a chloride salt hydrate, a co-crystal and a bulky flexible molecule. This blind test has seen substantial growth in the number of participants, with the broad range of prediction methods giving a unique insight into the state of the art in the field.
Publisher’s copy
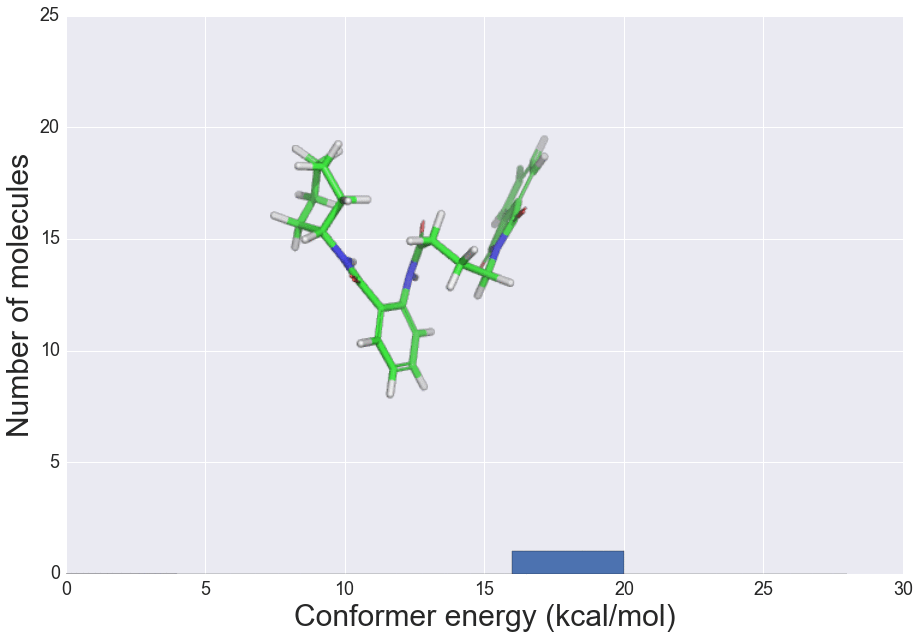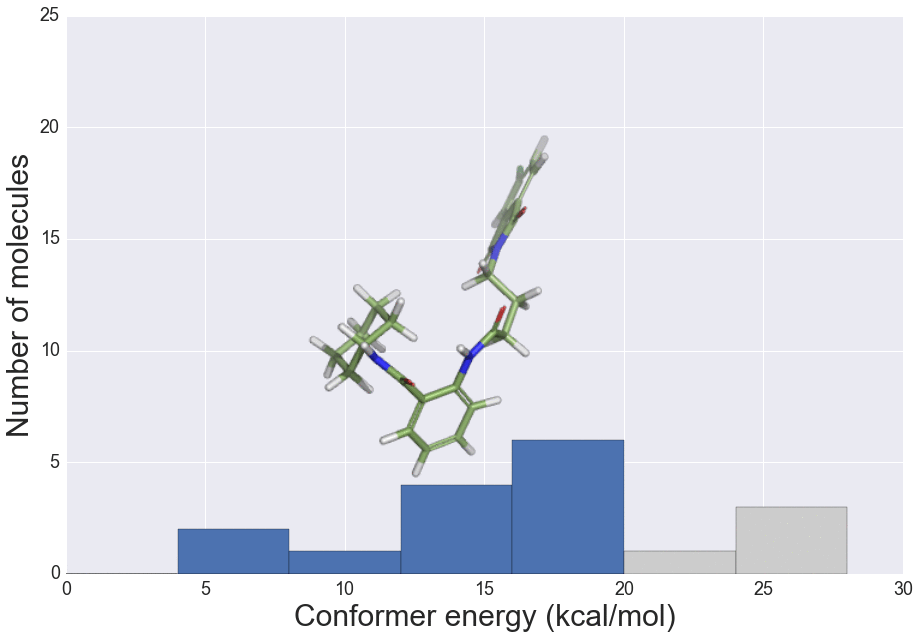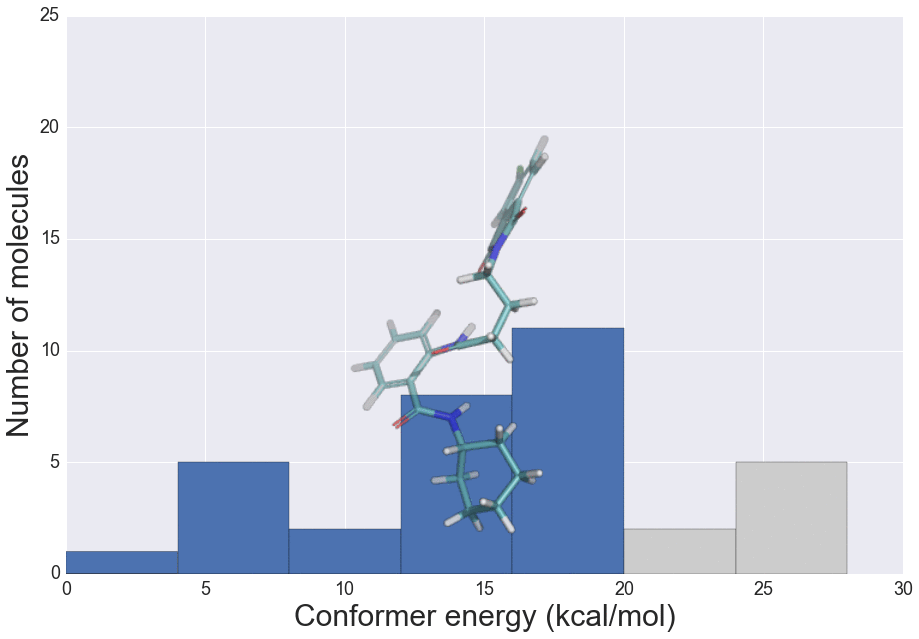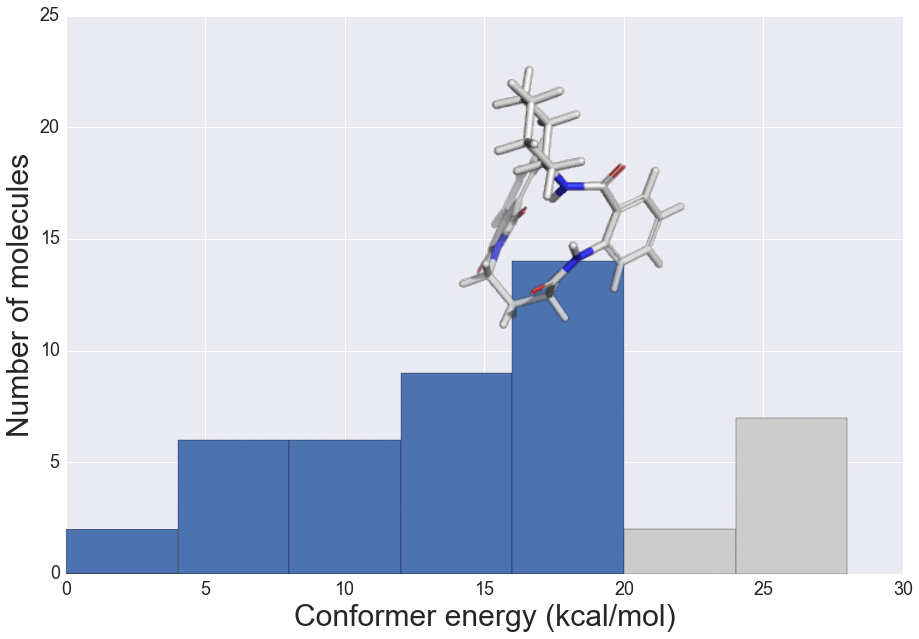 J. Chem. Inf. Model., 2016, 56 (12), 2347–2352 doi: 10.1021/acs.jcim.6b00565
J. Chem. Inf. Model., 2016, 56 (12), 2347–2352 doi: 10.1021/acs.jcim.6b00565