The CRYSTALS v6720 installer is now available.
The CRYSTALS v6720 installer is now available.
This update uses a new 64-bit Fortran compiler, fixes a number of bugs and eliminates a few mysterious crashes. Please report problems. The version number now refers to a specific snapshot of the source code enabling us to better identify and fix bugs.
Key changes between v14.6237 and 14.6720
64-bit compiler, bundle mkl math libraries – improved stability. Updated icon.
Improved SHELX import for atoms on special positions, SADI, DFIX and MPLA restraints, long form factors.
Spot is_nan in plot data.
New set of displacement parameter restraints, including TLS.
Better #CHECK output for restraints – including individual leverages.
Fix misreport of symmetry operators in CIF for C2/m
Dynamic threshold of Q peak height in Fourier peak viewer script.
Allow paths containing single quotes.
Add greek symbols to text and graphs for more concise display.
Leverages calculation via #sfls, refine punch=leverages, end
Changes related to getting consistent results for Thmax in CIF
Fixed listbox operation clicking in list now generates appropriate feedback for scripts.
Least squares matrix condition number output now included for problem refinement diagnosis.
Avoid almost infinite loop looking for bonds if atom coords have gone bad. Side effect – bonds outside +/- 50 unit cells of the origin won’t be found.
New choice of matrix invertors for SFLS. Default can be set in L23.
Fix default hkl format when re-reading ‘from-cif.hkl’ files (3F4.0,2F10.0).
Add a few more instrument/diffractometer types (synchrotron, neutron sources, etc.)
Modify platanom calculation: (i) use more d.p for improved precision; (ii) use c and d terms to correct data with all anomalous scattering effect removed as detailed in final version of ‘HUG and SQUEEZE’ manuscript.
Compute dispersion correction terms and photon interaction cross section for non-standard (=Mo,Cu) wavelength data. Alter xinlist3 to streamline experience.
Undo dialog: fix situation where current L5 is marked as error list.
Check for isotropic / unknown atoms before attempting to SPLIT in xsplits.ssc
Make L11 depend on L28, now if L28 filters change the VCV becomes invalid, require re-refinement. Avoids publishing CIF after omitting reflections without extra cycles.
Fix storage of converge checkbox state so it isn’t overridden when called from xwrite5.
Edlist3: revamp user interface to simplify. Added get/set mu values from L29 for each element.
Add Helium to properties file.
LIST 6 version incremented to 124. NEW DSC files will not be backwards compatible with older versions of CRYSTALS.
Better treatment of twins in Fourier maps.
Fix output of RIDE constraints (better refinement stability).
Reduce decimal places for angles with no esd (e.g. by symmetry constraint, or not refined) from 3 to 2 in CIF output. Avoids reporting e.g. 179.995 degress for exactly 180 degree angles.
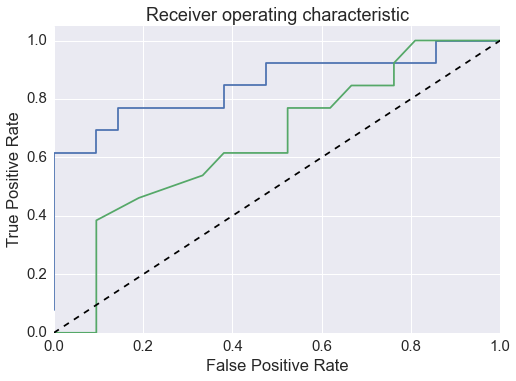
Add internal vs external variance plot
Fix switch on of extinction from the Guide ‘Add extinction parameter’ dialog.





 The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.
The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.